GB/T 40111-2021 石油产品中氟、氯和硫含量的测定燃烧-离子色谱法
警示一使用本文件的人员应有正规实验室工作的实践经验。本文件的使用可能涉及到某些有危险的材料.设备和操作,本文件并未指出所有可能的安全问题。使用者有责任采取适当的安全和健康措
施,并保证符合国家有关法规规定的条件。
范围
本文件规定了采用燃烧离子色谱法测定石油产品中氟、氯和硫含量的试验方法、干扰、仪器设备、试剂与材料.取样、准备工作.校准.试验步骤.结果计算.结果表示,以及楷密度和偏差。
本文件适用于沸点范围为30C~380C,测定范围为0.30mg/kg~15.00mg/kg的石油产品中氟、氯和硫含量的测定,包括汽油.乙醇汽油.石脑油.馏分油、喷气燃料.柴油、生物柴油调合燃料等。超
出此范围的样品.也可使用本方法进行测定,但是尚末确定其精密度。
2规范性引 用文件
下列文件中的内容通过文中的规范性引用面构成木文件必不可少的条款。其中,注H期的引用文件,仪该H期对应的版本适用于本文件;不注H期的引用文件,其最新版本(包括所有的修改单)适用于
本文件。
GB/T1884原油和液体石油产品密度实验室测定法(密度计法)
GB/T 1885石油计量表
GB/T 4756石油液体手 工取样法
GB/T 6682- -2008 分析实验室用水规格和试验方法
GB/T 27867石 油液体管线自动取样法
SH/T0604原油和石油产品密度测定法(U形振动管法)
3术语和定 义
本文件没有需要界定的术语和定义。
4原理
将试样注入高温燃烧管内,试样在高温.富氧、水解的条件下燃烧,燃烧产生的气体被收集在装有吸收液的吸收管内。在吸收管内,燃烧过程中产生的岗化氢(HX)在吸收液中生成岗素离子(X- );吸 收液
内含有的氧化剂将气体中硫的氧化物(SO。)进一步氧化生成硫酸根离子(SO,-)。仪器通过进样阀自动将吸收液定量注人到离子色谱单元,卤素离子(X )和硫酸根离子(SO,2 )经分离柱分离,用电导检.
测:器进行检测。通过外标法可得到试样中氟、氯和硫的含量,测定结果以质量分数表示。
5干扰
5.1与待测阴离 子共同洗脱出的其他阴离子(如:溴离子、硝酸根离子等)可能会对测定产生干扰。选择合适的淋洗液及淋洗液浓度可消除此干扰。

5.2某个阴离子含量较高时,可能会对与其保留时间接近的待测阴离子有影响,从面产生干扰。如:氟离子含量较高时,其洗脱时间较长,可能会对氢离子产生干扰。
6仪器设备
警示一高温,在燃烧炉附近使用易燃品时应特别小心.
6.1燃烧离子色谱仪
由燃烧单元.吸收单元和离子色谱单元三部分组成,其结构示意图见图1.
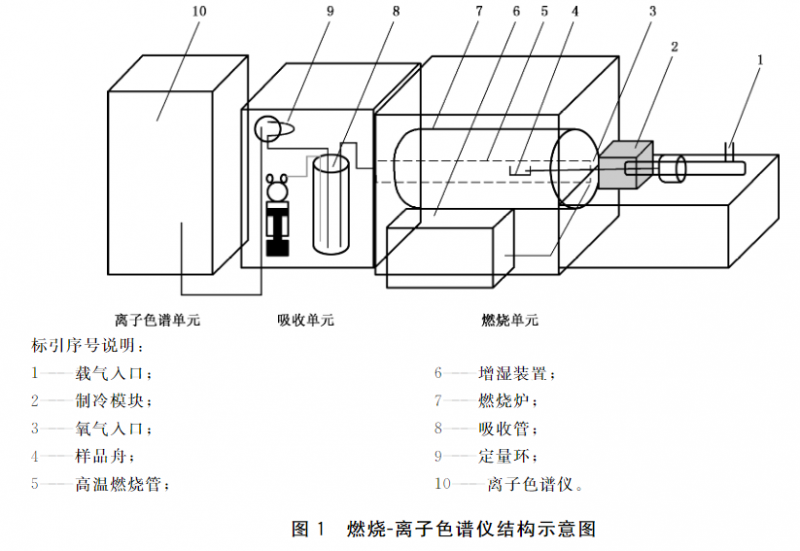
6.2 燃烧单元
6.2.1注射器:为 10 μL.25 μL,50 μL,100 L或250 μL。进样方式可采用自动进样器进样,也可采用手动方式进样。
注:试样分析完毕,准备分析下一个试样时,宜彻底清洗注射器。先用待测离子含量低的溶剂清洗,如:配制标准I作溶液时用的溶剂,再用下一个试样反复冲洗。或按厂家的建议设定清洗步骤,以避免样品间交叉污染。
6.2.2舟进样系统:结构见图2。 系统上有一个进样口,与高温燃烧管人口相连;前端有载气入口,整个系统用载气吹扫;系统有一个可控进样速度的进样器,可将装有试样的样品舟按指定速度推至高温燃烧
管内,也叮将样品舟从高温燃烧管内拉出。从高温燃烧管拉出的样品舟退至进样口处,此处有制冷模块(6.2.3),可使样品舟完全冷却。待样品舟完全冷却后可进行下一次样品的测定。
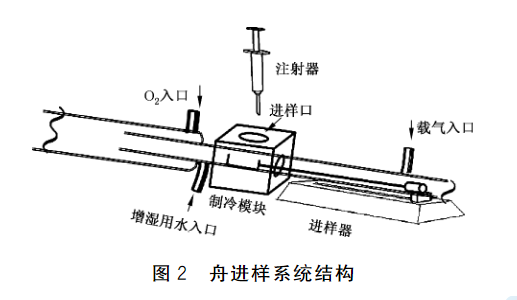
6.2.3 制冷模块:宜使用电子制冷模块。其主要作用是确保试样被注射至样品舟时,样品舟已经完全冷却,防止试样挥发。
6.2.4流量控制器:为确保氧气和载气流速稳定,仪器应配置气体流量控制器。
6.2.5燃烧炉:控制温度不低于900°C,精度为土25C。所设温度应确保全部试样能完全氧化燃烧。
6.2.6 高温燃烧管:石英材质,可耐1 100 C高温。高温燃烧管空间应足够大,以确保试样能够充分地燃烧,为提高试样的燃烧空间和接触面积可加入石英毛(7.13)或者其他适合的物质。高温燃烧管有两
个侧臂,分别为增湿用水人口管线和氧气管线,主要是为了达到富氧和水解的燃烧环境。
6.2.7增湿装置:可将-一定流速的一-级水(7.1)通人高温燃烧管中,从而得到高温水解的环境。
6.3吸收单元
6.3.1 连接管:用于连接高温燃烧管和吸收管,应有人水口和出水口的管线。试样在高温燃烧管中完全燃烧后,仪器自动地用一-级水(7.1)冲洗连接管,并全部转移至吸收管(6.3.2)内。
6.3.2 吸收管:至少装10 mL吸收液。在样品燃烧过程中,管内的吸收液连续吸收气体及全部液体,以确保所有的燃烧产物(HX和SO,)均被吸收。燃烧结束后,可自动定容至所需体积,并通过定量环自动
注射定量的吸收液到离子色谱仪器中。
6.3.3吸收液被注人到离子 色谱仪器后,且在下一个试样燃烧前,用一-级水(7.1)冲洗连接管及吸收管,以尽量减少前一个样品的污染。
6.4 离子色谱单元
6.4.1泵:输送淋洗液。
6.4.2 连续淋洗液发生器(可选):可自动制备和净化淋洗液的装置,包括电解淋洗液发生器法和自动定量管法。在不降低方法精密度和准确度的前提下,可使用其他连续淋洗液发生装置。
6.4.3阴离子预浓缩柱(可选):主要起到预浓缩阴离子和消除基体效应的作用。如果安装了预浓缩柱,则可在吸收液注人保护柱和分离柱之前去除其中剩余的过氧化氢,从而消除对氟离子色谱峰分离度
的潜在干扰。
6.4.4保护柱:保护分离柱免受强保留成分污染。同时还可获得更好的分离效果。
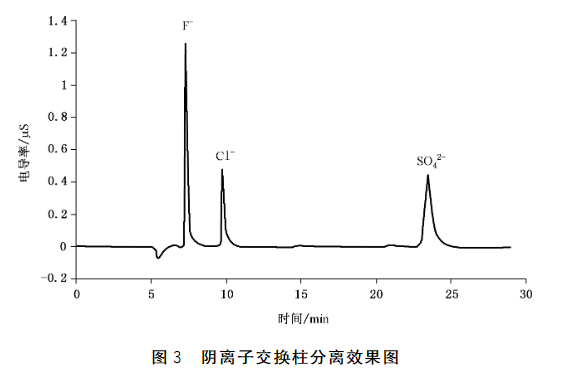
6.4.5分离柱:阴离子交换柱,可使待测物的阴离子峰达到基线分离的效果,如图3所示。
6.4.6抑制器:可选择化 学抑制器或连续电解抑制器,其作用是降低通过分离柱后淋洗液的背景电导率。在不降低方法精密度和准确度的前提下,也可使用其他抑制器。
6.4.7检测器:选用电导检测器。
6.4.8数据采集系统:可对离子色谱峰进行积分的积分仪或电脑数据处理系统。
6.5样品舟
材质为石英或陶瓷。样品舟填满石英毛(7.13)或其他合适的材料,以吸附注射器针头上残留的试样。
6.6分析天平
感量为0.1 mg。
6.7容量 瓶
规格为100mL.1000mL和2000mL。
7试剂与材料
警示一氢氧化钾是苛性碱,防止接触眼睛、皮肤等身体部位。过氧化氢是强氧化剂,防止接触眼睛、皮肤等身体部位。
试验过程中所使用的试剂均为分析纯,在不降低测定结果精密度的前提下,可使用其他纯度的试剂。
7.1 一级水:应符合GB/T 6682- -2008 中一-级水的要求;氯含量不大于1 μg/L;25 C时电阻率大于18 Mn●cm;总有机碳小于501rg/L。
7.2 对氟苯甲酸:相对分子质量140.11;氟理论质量分数为13.56%。
7.3 氟苯:相对分子质量96.10;氟理论质量分数为19.77%。
7.4 2,4,5三氯苯酚:相对分子质量197.46;氯理论质量分数为53.87%。
7.5 氯苯:相对分子质量112.56;氯理论质量分数为31.50%。
7.6 二米并噻吩:相对分子质量184.26;硫理论质量分数为17.40%。
7.7 30% 过氧化氢:其中每个待测离子浓度均宜小于1.00 mg/kg。
7.8淋洗液:氢氧化钾溶液,或碳酸钠/碳酸氢钠溶液。在不降低测定结果精密度的前提下,也可使用其他淋洗液或购买合适的市售溶液。
7.9抑制液:使用化学抑制器时需要使用,按操作说明要求配制、使用。电解抑制器只需根据淋洗液浓度按仪器推荐值设定电流值即可。
7.10 溶剂:异辛烷、二甲苯、甲苯或与待测样品组分相似的其他溶剂,用于溶解溶质和稀释样品。溶剂中待测元素含量应小于0.05mg/kg。每瓶溶剂空白均应检测。
7.11 吸收液:取0.7 mL 30%过氧化氢(7.7)于2 000 mL容量瓶中,用一-级水(7.1)稀释至刻度。 吸收液中加入过氧化氢是为了使待测气体中的SO。全部转换成SO,2-。如果不测定样品中的硫含量,则吸收液可不加入过氧化氢,使用一- 级水(7.1)作为吸收液。
注1:满足方法精密度的前提下,也可使用其他浓度的吸收液。
注2:样品中硫含量低于15mg/kg时,可不加入过氧化氢。具体根据仪器测定情况进行选择,当吸收液中加与不加过氧化氢所得到的硫峰面积计数差别小于5%时,则吸收液中可不加入过氧化氢。
7.12质量 控制(QC)样品:稳定的、具有代表性的样品或使用标准工作溶液(9.2.2)。用于验证整个试验过程的准确性,见第14章。
7.13石英毛。
7.14 载气:氩气或氦气,纯度不低于99.99%。
注:可用净化装置去除杂质,如:川分子筛去除水气;用活性炎或其他具有同样功能的物质吸附烃类化合物。
7.15氧气:用为燃烧气 ,纯度不低于99.75%。
7.16氮气(可选):通 入淋洗液罐,防止空气进人或产生气泡,纯度不低于99.99%。
8取样
8.1按GB/T4756和GB/T27867方法取样。含有易挥发性的组分的样品应在测定前打开装样容器。取出样品后应尽快分析,避免造成测定元素的损失和污染。
8.2样品采到容器后,如不立刻使用,则应在分析前将样品在容器内充分混匀。
注:由于样品中存在轻组分,所以混匀时要注意安全。
9准备工作
9.1仪器的准备
9.1.1按仪器说明书要求安装仪器。
9.1.2
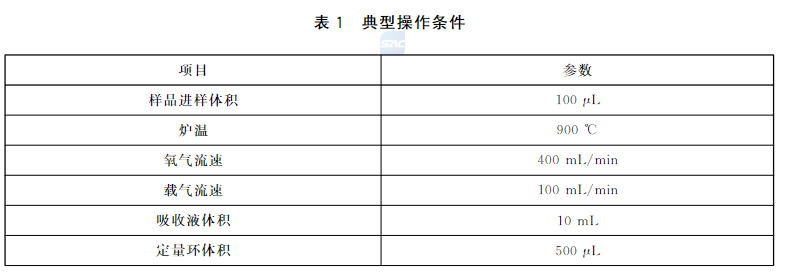
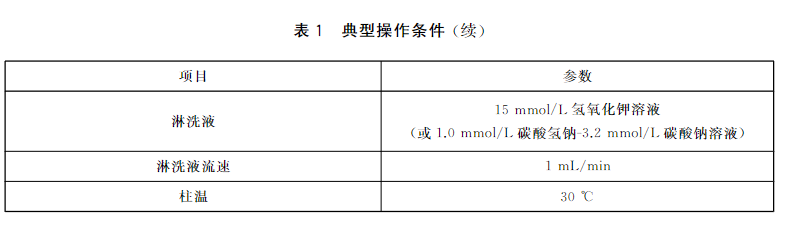
调节仪器的操作参数。表1给出了典型操作条件。
9.2标准溶液的配制
9.2.1 标准储备液(100 mg/L):可购买市售的能够满足使用要求的标准储备液;也可按下述方法进行配制。分别准确称取约0.737 5 g对氟苯甲酸或0.5059 g氟苯、0.185 7 g 2,4,5三氯苯酚或0.3174 g氯苯和0.574 8 g二苯并噻吩至1 000 mL容量瓶中,再用所选溶剂(7.10)稀释至刻度。标准储备液可进一步稀释至标准工作溶液的各个浓度。标准储备液质量浓度按式(1)计算。

注:配制标准工作溶液时,宜对溶质的化学杂质进行校正。
9.2.2 标准工作溶液:可购买市售的能够满足使用要求的标准工作溶液;也可由标准储备液进一步稀释得到不同浓度的溶液,用于建立校准曲线。具体配制浓度见10.2。
注1:根据标准储备液、标准工作溶液使用频率和有效期,需定期配制。如果冷藏,--般有效期为3个月,如果选用苯为溶剂时,则不能冷藏,因为苯在低温时会结晶,导致标准溶液稳定性差。
注2:标准储备液、标准工作溶液也可采用重量法配制。
10 校准
10.1 分析标准工作溶液前,需进行-系列系统空白和溶剂空白的测定,以确保系统是干净的。宜采取.如下操作:
a)测定系统空白:直接将未注人任何试样或试剂的样品舟推人高温燃烧管中,并按分析试样时的.条件进行分析。反复测定系统空白,直到仪器基线稳定,并且连续三次测定的待测离子系统空
白峰面积的相对标准偏差不大于5%,此时可认为得到了一个稳定的系统空白。如果无法得到稳定的系统空白,应查找污染源。反复测定系统空白,使系统空白的峰面积不大于溶剂空白
峰面积的50%。
b)测定溶剂空白:向样品舟内注入配制标准工作溶液时所用溶剂,并按测定试样时的条件进行分析。反复测定溶剂空白,使溶剂空白的峰面积不大于校准曲线上所用标准工作溶液最小浓
度峰面积的50%。
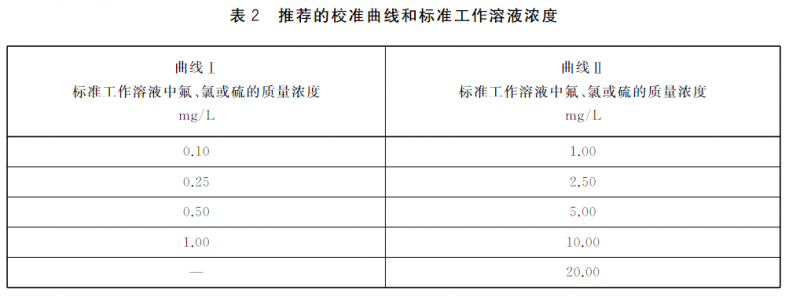
10.2根据待测样品含量,购买或者配制系列标准工作溶液,标准工作溶液的浓度应能涵盖待测样品的浓度。表2给出了推荐的校准曲线和标准工作溶液浓度。在精密度能达到方法要求的前提下,也可使
用其他浓度的标准工作溶液建立校准曲线,但应包括待测样品的浓度。
10.3在选定的测定条件下,分析每个标准工作溶液,得到每个标准工作溶液的色谱图。以标准工作溶液中待测元素的色增峰面积为纵坐标,相对应的质最浓度为横坐标,按仪器说明书建立各个元素含量与峰面积的校准曲线。每个元素典型的校准曲线应至少包括4个不同浓度的标准工作溶液,并且应涵盖待测元素的浓度。校准曲线的相关系数(r2)应大于0.995。
10.4分析标准工作溶液后,检查高温燃烧管和气体所经的流路中各个部件是否存在积炭,以确保标准工作溶液完全氧化燃烧。如果发现样品舟中有积炭生成,应增加样品舟在燃烧炉内的停留时间。如果在高温燃烧管的末端发现有积炭生成,则应减缓样品舟的进样速度或增加样品舟在高温燃烧管前端的停留时间,也可同时采取这两种措施。按仪器说明书要求清除部件上的积炭,重新组装仪器。
10.5标准工作溶液分析完成后,样品舟被拉回制冷模块(6.2.3)上。在制冷模块上样品舟应至少停留100s,或更长停留时间,使样品舟能完全冷却,然后再进行下--个标准工作溶液的测试。
11试验步 骤
11.1 按第8章准备样品。
11.2用注射器将定量的试样,如100pL,注人到样品舟中;按分析标准工作溶液的测定条件,以同样的方式测定试样,得到试样的色谱图;对氟、氯和硫元素的色谐峰进行积分,得到各元素色谱峰面积。为了减少燃烧效率、吸收效率等带来的误差,分析试样的条件应和分析标准工作溶液条件--致。
11.3分析试样后,检查高温燃烧管和气体所经的流路中各个部件是否存在积炭,以确保试样完全氧化燃烧。如果发现样品舟中有积炭生成,应增加样品舟在燃烧炉内的停留时间;如果在高温燃烧管的术端发现有积炭生成,则应减缓样品舟的进样速度或增加样品舟在高温燃烧管前端的停留时间,也可同时采取这两种措施。按仪器说明书要求清除部件上的积炭,重新组装仪器。
11.4 试样分析完成后,样品舟被拉回制冷模块(6.2.3)上。在制冷模块上样品舟应至少停留100 s,或更长停留时间,使样品舟能完全冷却后,再进行下- -个试样的测试。此步骤对于初馏点低的试样尤为
重要。
注:如果样品舟的冷却时间短,可能会导致轻组分试样挥发.生成积炭.甚至有明能发生爆燃的现象.
12结果计算
12.1如果离子色谱仪自带计算机数据处理钦件.则用试样的各个元素的峰面积在校准曲线上自动计
算出试样中氟、氯和硫元素的质量浓度。如果离子色谐仪没有计算机数据处理软件,可通过峰面积和校
13结果表示
取重复测定两次结果的算术平均值作为测定结果,单位为毫克每千克(mg/kg),结果保留至0.01 mg/kg.
14 质量控制
14.1 每次开机后,在分析前至少应对QC样品(7.12)进行- -次测定。
14.2若实验室建立了质量控制及质量保证程序,可按其确认测定结果的可靠性。
14.3 若实验室未建立质量控制及质量保证程序,可参考NB/SH/T 0843作为评价系统。
15精密度和偏差
15.1 精密度
15.1.1 概述
精密度通过6个实验室对22个样品,包括:汽油、乙醇汽油、柴油、石脑油、馏分油、喷气燃料、柴油、生物柴油调合燃料等进行协作试验得到,并参照GB/T 6683通过实验室间统计分析结果确定。按
15.1.2和15.1.3 判断试验结果的可靠性(95%置信水平)。
注:如果没有妥善保存含有易挥发组分的试样,则会影响其测定结果。
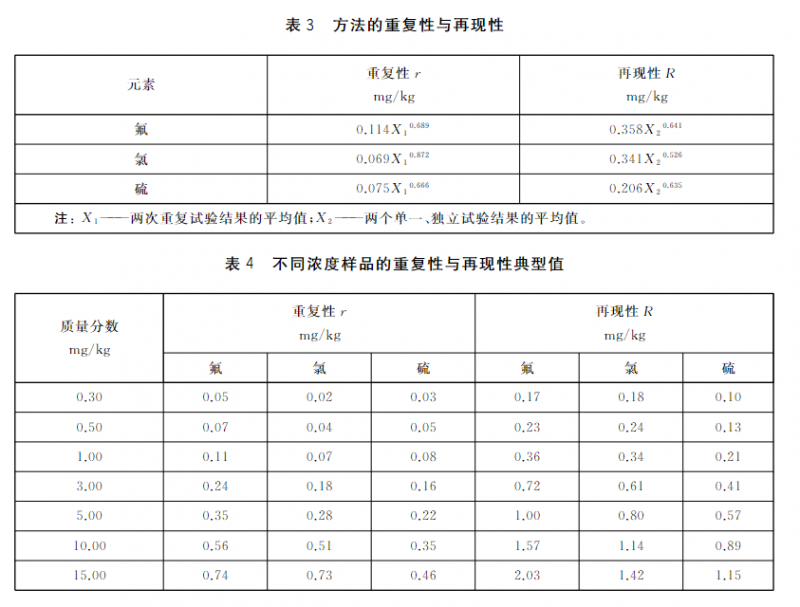
15.1.2重复性(r)
同一操作者,在同一实验室,使用同--仪器,对同一试样进行连续测定,所得两个试验结果之差不应超过表3中方法的重复性限值。不同浓度样品的重复性典型值见表4。
15.1.3 再现性(R)
不同操作者,在不同实验室,使用不同仪器,按相同的试验方法,对同一试样分别进行测定,所得两个单一、独立的结果之差不应超过表3中方法的再现性限值。不同浓度样品的再现性典型值见表4。
15.2 偏差
由于没有可用于测试本方法偏差的参考物质,故本文件未给出偏差。
参考文献
[1]GB/T6683石油产品试验方法精密度数据确定法
[2] NB/SH/T 0843 石化行业分析测试系统的评价 统计技术法
15.2ÚÛO¶üh?¶!³ ́0MÅ?,!"#ÒN_³ ́。15.2ÚÛO¶üh?¶!³ ́0MÅ?,!"#ÒN_³ ́。




 微信客服
微信客服



